| Project Leader: Prof. Jan Augustyński | Project period: 2014 - 2020 |
| Project funding: MAESTRO 5, NCN | |
| Project description: Among the fields in which thin-layer oxide materials find application one can mention chemical sensors, electrocatalytic electrodes (such as those used in fuel cells) and photo-electrochemistry. The planned project intends to explore the domain situated among those two latter fields. The proposed extensive combinatorial investigation of thin film semiconducting metal oxide materials, including various doped binary oxides and ternary oxides is expected to allow discovery of new materials with significantly improved properties such as optical absorption range, charge-transport and charge-transfer. While the first characteristic is of primary importance for the application of photo-materials for water splitting or photo-oxidation of chemical effluents, both latter properties are of major interest also in the field of electrocatalysis. The research conducted within the Maestro project covers a variety of metal oxide materials, either (i) semiconductors or (ii) exhibiting metallic conductivity. The first category is intended for application as photo-anodes in photo-electrochemical (PEC) devices whilst in the case of the second category we are looking for particular electro-catalytic properties as anodes in the absence of illumination or also in conjunction with the semiconductor photo-electrodes. Since the very beginning, on the basis of our earlier experience, we chose the approach of thin layer, nano-crystalline semiconductors that combine high photoactive surface area and large porosity allowing permeation of most of the photo-electrode by the electrolyte. Since none of the oxide semiconductor photo-anodes, investigated till now, are able to directly split water (i.e., to produce hydrogen) in the presence of a metal cathode under visible light, our search of novel materials and their structure was governed by the requirement to make them able to operate in a tandem device. This concept developed earlier in the author’s laboratory (J. Mater. Chem., 2008, 18, 2298) consists in combining in series two optical systems: the photo-electrolysis cell and a photovoltaic (PV) cell, placed behind, intended to provide the bias voltage required to split water in the first cell. The single-junction PV cell captures the complementary parts (longer wavelengths) of the solar spectrum transmitted through the photo-electrolysis cell. To be viable this concept requires thin-layer, semi-transparent photo-anodes attending saturation photo-current under a bias voltage not exceeding ca 1 V that can provide a dedicated PV cell. Figure 1. Schematic representation of a twin PEC/PV device (tandem cell) used in water splitting under solar light irradiation. In this connection, the important result of the first phase of the project was the demonstration of very high, reproducible water splitting photo-currents of 4.5 mA cm-2 obtained under simulated 1 sun AM 1.5G irradiation with nano-crystalline (NC) tungsten oxide (WO3) photo-anodes polarized at ca 1 V versus hydrogen electrode (RHE). This performance was due to the extension of visible light absorption by moderate doping of WO3 with sodium ions and to the use of Keggin-type polyoxometalate (POM) oxygen evolution reaction (OER) catalyst in solution, the small amount (10-4 mol/L) of which is added to the electrolyte. We postulated that the presence of highly hydrated POM anions adsorbed within positively charged (at pH 0) WO3 film leads to a decrease of reorganization energy of water oxidation reaction. Another important advantage of POMs is their high proton conductivity facilitating removal of H+ ions from the reaction site after consecutive discharges of water molecules. Importantly, this is the first example of an effective OER catalyst stable in acidic electrolytes that does not include noble metals. In view of the implementation in a tandem cell, the large, stable water splitting photo-currents attained with the developed (Na)WO3 photo-anodes translate into chemical (hydrogen)-to solar efficiency exceeding 5% (Adv. Energy Mater. 2016, 6, 1600526). In fact, a relatively low anodic bias at which the photo-anode reaches a saturation photo-current should allow autonomous operation of a tandem device
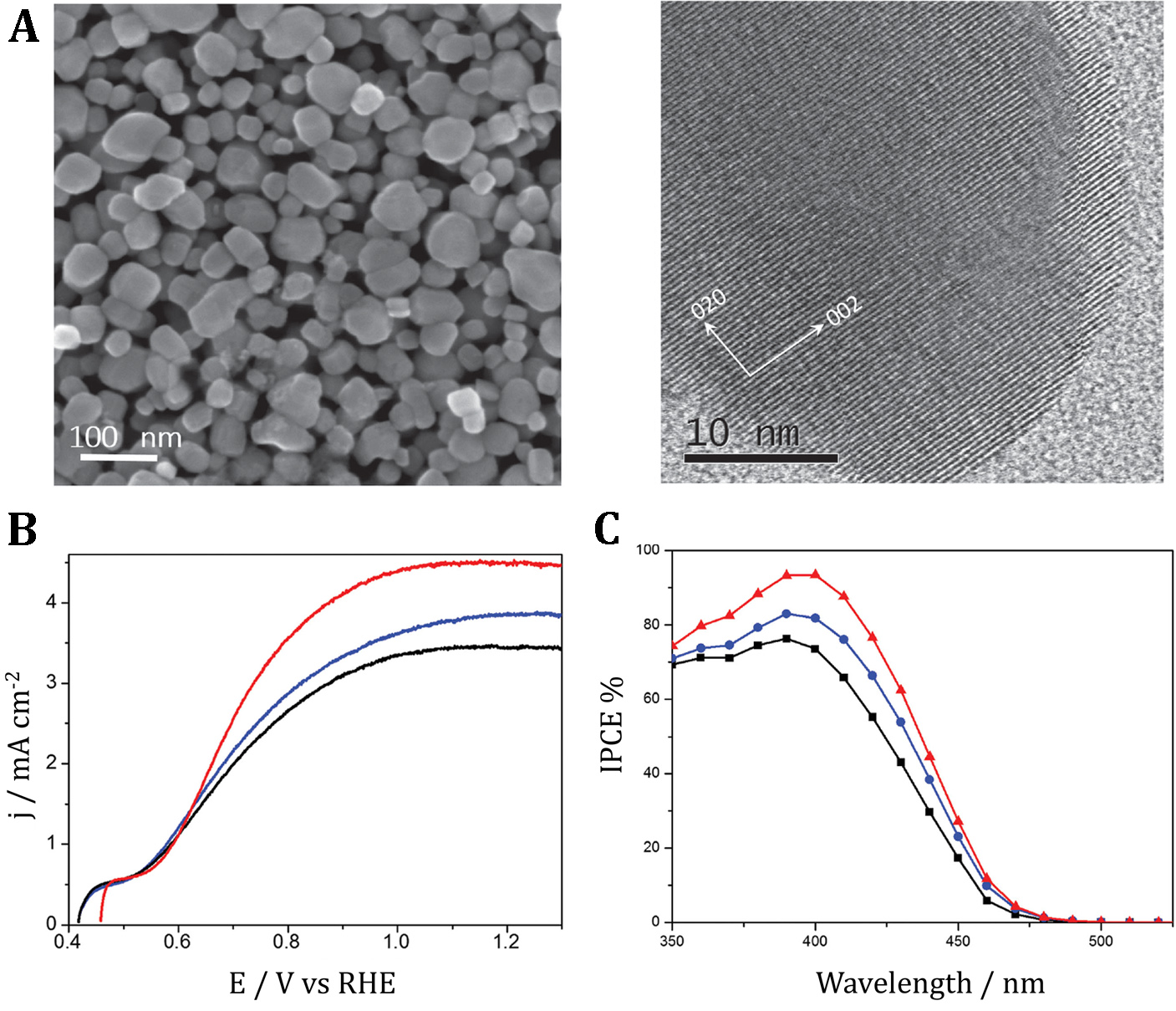
Figure 2. A) Scanning electron microscope (SEM, left) and transmission electron microscope (TEM, right) images of a WO3 film obtained by a sol–gel method after final annealing at 550°C. B) Current–potential (j–E) curves for ca ≈2.5-μm-thick WO3 photo-anodes: Nominally undoped WO3 (in black) and 0.4 at% Na-doped WO3 (in blue) recorded in a 1M CH3SO3H solution and, for the latter electrode, after addition to the electrolyte of 10−4M of H3PMo12O40 (in red): Simulated AM 1.5G irradiation. C) IPCE plots recorded for 2.5-μm-thick nominally undoped (in black) and 0.4 at% Na-doped (in blue) WO3 photo-anodes in 1M CH3SO3H electrolyte and for the (Na)WO3 photo-anode in 10−4M of H3PMo12O40 /1M CH3SO3H (in red). The improved (Na)WO3 photo-anodes were also used in a solar-driven cell using an electron-coupled proton buffer, phosphomolybdate, (PMo12O40)3- . In the course of the photo-electrolysis proceeding without any external bias, in parallel with oxygen evolution occurring at the photo-anode, the phosphomolybdate is reduced into (PMo12O40)5- at the cathode. The latter may be eventually stored and subsequently used in the second, electrochemical re-oxidation step of the process to generate hydrogen. Among advantages of this concept, consisting in decoupling the process of water splitting in separate oxygen evolution and hydrogen formation steps, is the possibility to form hydrogen with much higher intensity than in a direct photo-electrochemical water splitting reaction. (J. Am. Chem. Soc., 2016, 138, 6707).
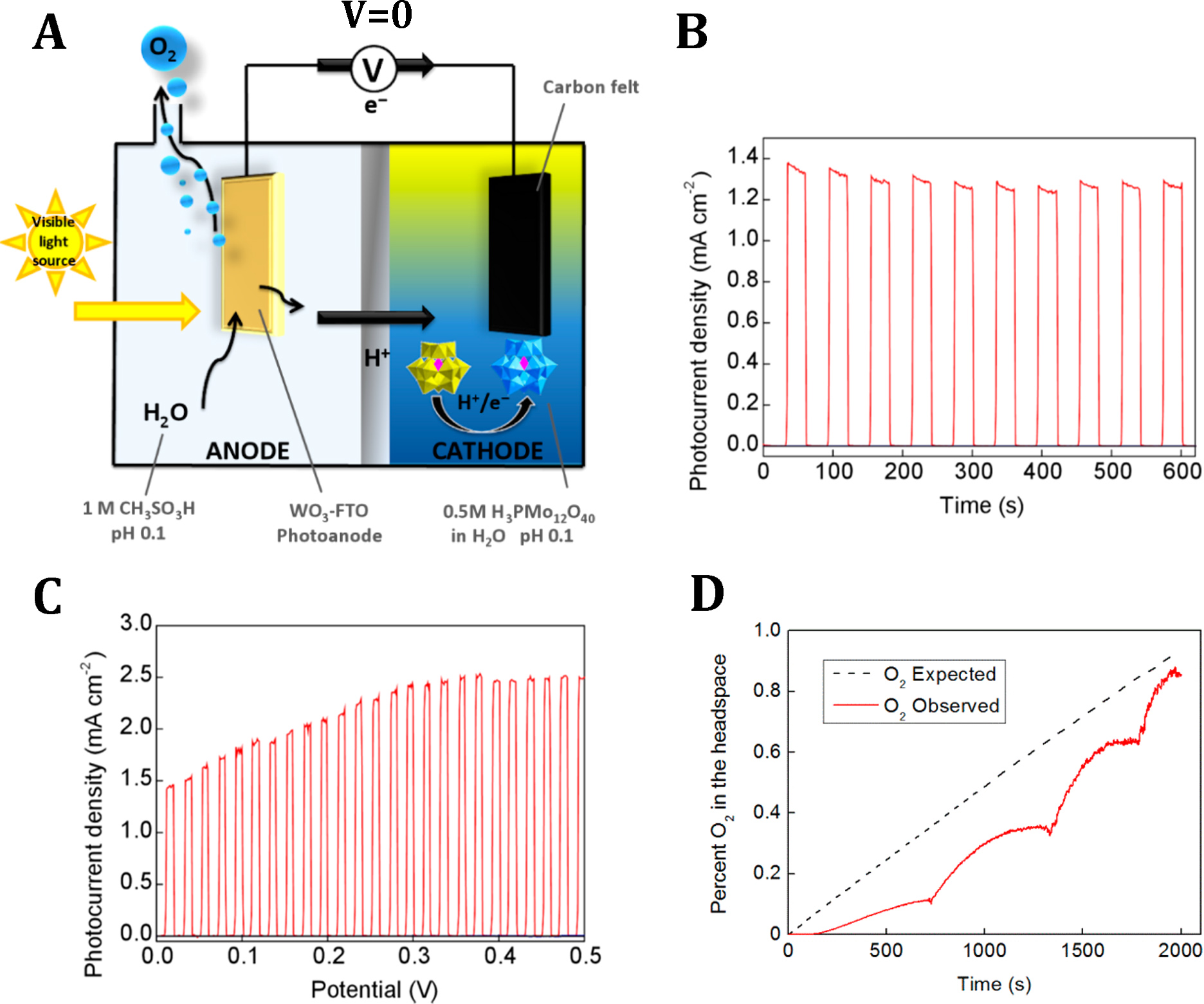
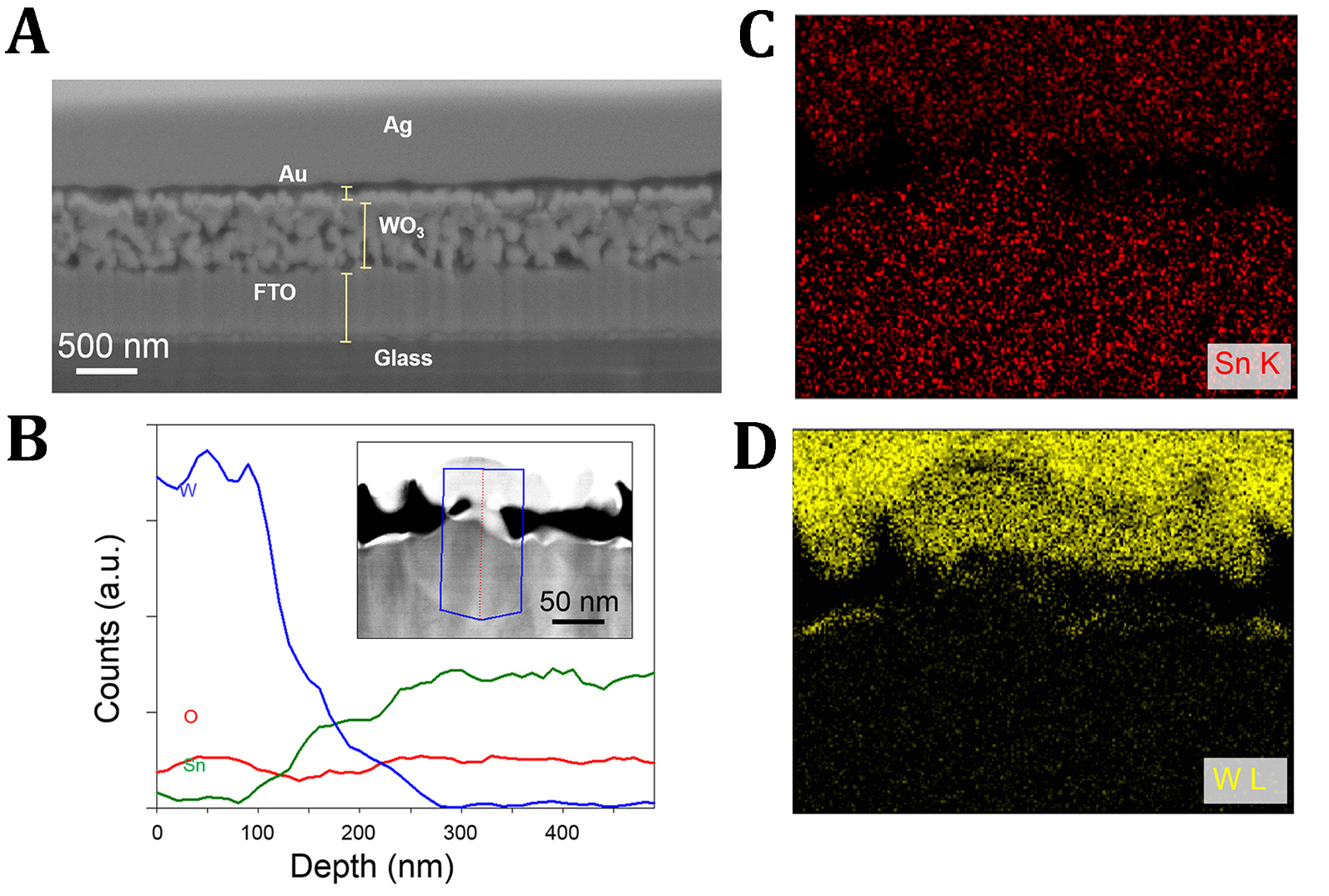
Figure 3. A) General schematic of the photo-electrochemical cell configuration used to drive water oxidation and concomitant reduction of the ECPB at zero bias. (B) Chopped light experiment in a two-electrode configuration at 0 V bias, according to the cell setup in Fig. 3A (red line), (C) Chopped light experiment in a two-electrode configuration, as above, but with the bias potential varied from 0 to 0.5 V at a rate of 1 mV/s. D) Comparison of the amount of O2 expected in the cell headspace on the basis of the charge passed (black dashed line) and O2 detected in the cell headspace by fluorescence-quench measurements (red line). Bubbles collected on the anode and were periodically dislodged by shaking, accounting for the increments observed in the measured O2 signal. Another important discovery, made along the following stage of the project, was the identification and characterization of nano-hetero-structures formed at the interface between the NC WO3 films and the tin oxide (F-SnO2) of the conductive glass substrate. As the consequence of the higher temperatures (up to 670°C) annealing of the thin WO3 films, we observed an unusually large increase of both optical absorption and water splitting photo-currents. Extensive spectroscopic (XPS, SIMS) and structural (STEM-EDX) investigations clearly indicated migration of Sn(IV) from the substrate into the nanoporous WO3 film as well as uneven tin distribution along the modified film. These investigations also allowed exclude WO3 in-doping to result from the high temperature annealing. On the other hand, STEM-EDX and other complementary analyses suggest that the migration of the tin species occurring along WO3 crystallites leads to the formation of nano-hetero-junctions that affect optical properties of the film through increased dispersion of the incident light and improve separation of photo-generated charge carriers within the photo-anode (ACS Catalysis 2018, 8, 10573).
Figure 4. STEM-EDX analyses of the cross section of a ∼0.25-μm-thick WO3 film after annealing at 670°C: (A) SEM image of the analyzed cross section, and (B) EDX line scans for W, Sn, and O, corresponding to the red line within the delineated fragment in the STEM image (shown <4 as an inset). Panels (C) and (D) show maps for elemental Sn and W.
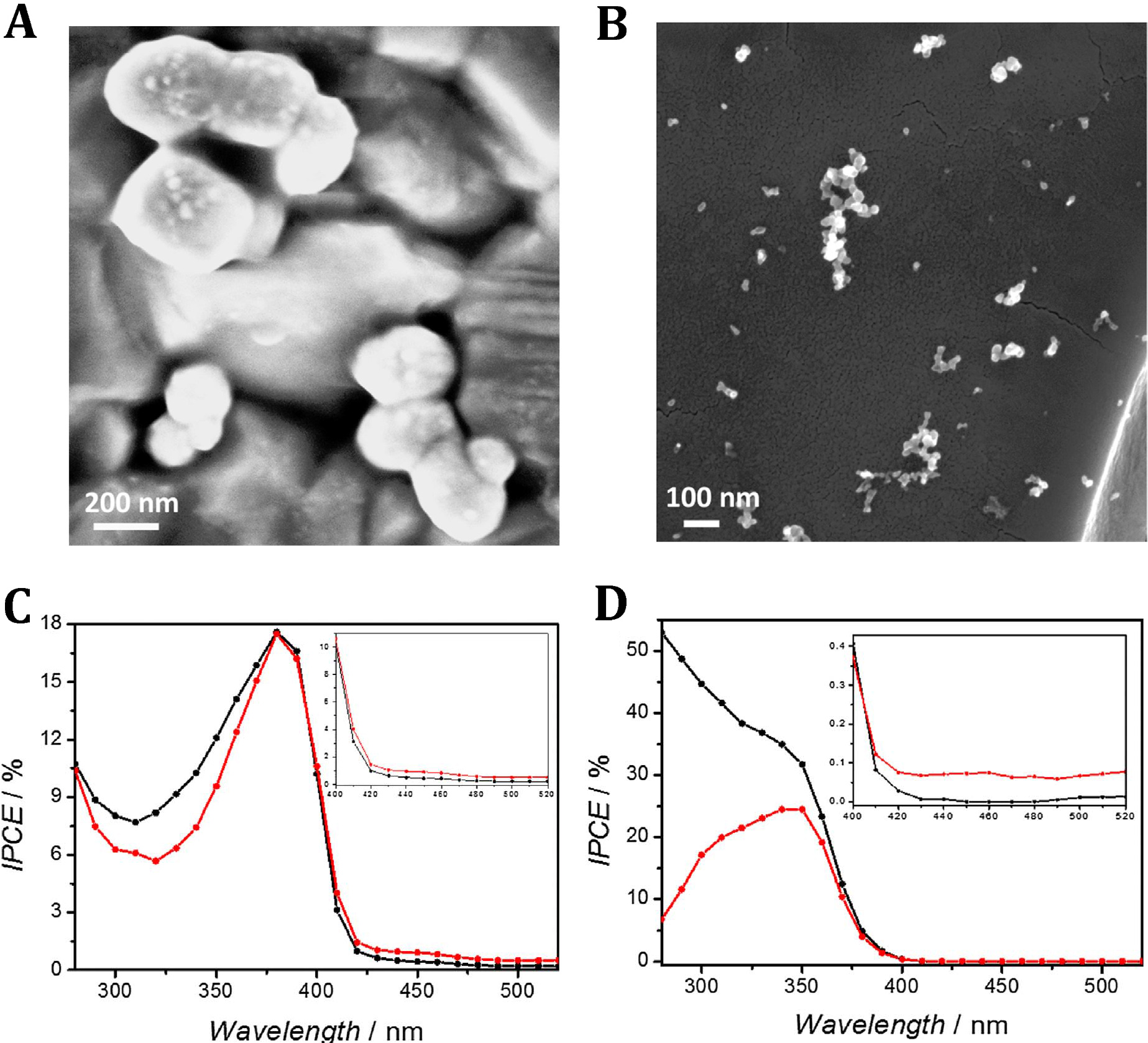
Figure 5. PEC water oxidation currents plotted against applied potential for WO3 electrodes annealed at 550°C (black curves) and after further annealing for 36 min at 670°C (red curves) for a 1.2-μm thick film. Measurements were performed in a 1M CH3SO3H supporting electrolyte under simulated AM 1.5G (100 mW cm−2) sunlight. IPCE photoaction spectra determined for a 1.2-μm-thick WO3 film annealed at 550°C (black curves) and further annealed at 670 °C (red curves). A significant part of our research dealt also with another n-type semiconducting material titanium oxide (TiO2) and its modifications. Following our earlier work on the enhancement of semiconductor’s (principally WO3) photoactivity by deposition of plasmonic metal – silver and gold nanoparticles (NPs), a novel study focused on Au/TiO2 and Ag/TiO2 photo-electrodes. Over the last decade, similar photocatalysts in the form of powder suspensions (mainly TiO2 decorated with Au NPs) were widely investigated with a view to degrade organic solution contaminants under visible light frequencies. The use of Au/TiO2 and Ag/TiO2 photo-electrodes opened the possibility to monitor, by recording the photo-currents, the changes in activity occurring in the course of photo-oxidation reactions of organic species in solution. Such measurements, performed in function of the light wavelength, provide incident photon conversion efficiencies (IPCEs) of the photocatalyst over the degradation reaction. Monitoring the IPCE spectra during a few hours long photo-oxidation of model organic molecules at a Au/TiO2 photo-electrode showed a drastic drop of IPCEs in the visible light range (420-600 nm) over which the gold NPs sensitize TiO2. At the same time, the IPCEs at the near-UV wavelengths (up to 420 nm) corresponding to the fundamental absorption range of rutile TiO2 remained unchanged. The deactivation of the Au NPs was assigned to their surface photo-oxidation covering the NPs with a Au(OH)3 shell and, thus, blocking the access of the organic species (J. Electrochem. Soc. 2017, 164, H667).
Figure 6. Scanning electron micrographs of a TiO2 – rutile electrode decorated with gold NPs; A) as prepared, B) after several sequences of photo-oxidation of acetic acid under irradiation with visible light (420–600 nm) wavelengths. C) IPCE spectra for the Au/TiO2 photo-anode collected during consecutive sequences of irradiation within the 420–600 nm range of wavelengths in 5 vol.% CH3COOH at 0.6 V vs. Ag/AgCl. The arrow shows the direction of changes in the IPCEs. The deactivation of the plasmonic NPs was further also confirmed during prolonged photo-oxidation of model organic compounds conducted at the anatase TiO2 electrodes decorated either with Au or Ag particles (Catalysis Today 2019, 321-322, 52).
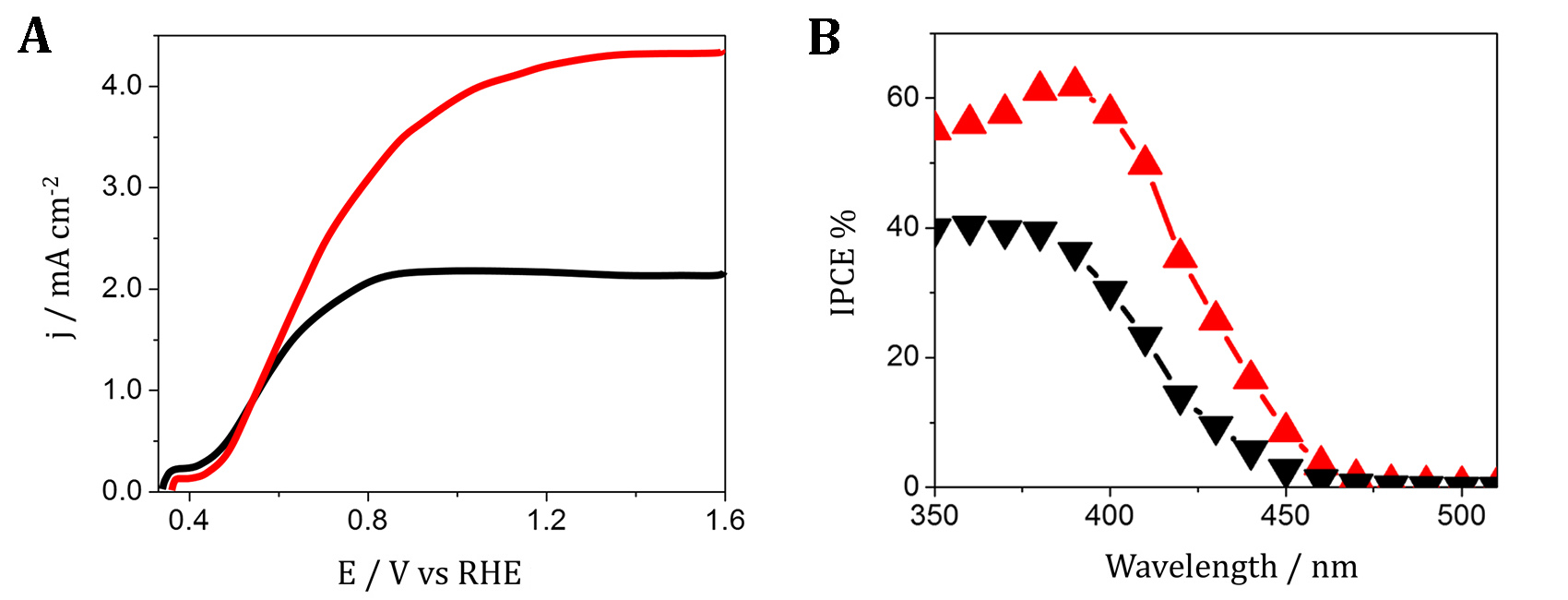
Figure 7. Scanning electron micrograph of electro-deposited (double-potential-pulse electrodeposition) silver-modified rutile (A) and anatase (B) electrodes. Incident photon-to-current conversion efficiency spectra recorded with rutile (C) and anatase (D) electrodes measured before (black) and after electrodeposition of silver particles (red), immersed in 0.1M NaCl with 5 vol.% CH3COOH. The inset shows expanded 420–600 nm region of the IPCE spectrum. Imposed potential: 0.4 V. Our research on visible light extension of the absorption range of TiO2 also involved preparation of various forms of sub-stoechiometric (defective) oxide. The principal difficulty met in these cases is to enable combination of improved visible light absorption with enhanced (e.g., in water splitting) photo-currents. A relatively simple method allowing formation of defective titanium oxide consists in the high-temperature (ca 850°C) oxidation of the Ti metal. However, despite the shift of the light absorption towards longer wavelengths, such films exhibit greatly reduced photo-currents (and IPCEs) within the fundamental absorption range of TiO2, suggesting strong recombination of photo-generated charge carriers occurring in the outer part of the formed defective film. Interestingly, we found that removal by etching of the most of the film thickness leads to the formation of highly photo-active electrode reaching IPCEs close to 40% at 400 nm and above 50% over near-UV wavelenghts. As showed by the established Mott-Schottky plots, another consequence of the activation of such films is a very large increase (by two orders of magnitude) of the concentration of charge carriers and the negative shift of the flat-band potential. Careful XRD measurements allowed identification in those defective titanium oxide films of the lower Ti3O oxide indicative of the migration into the film of titanium from the substrate (Solar Energy Mater. Solar Cells 2020, 208, 110424).
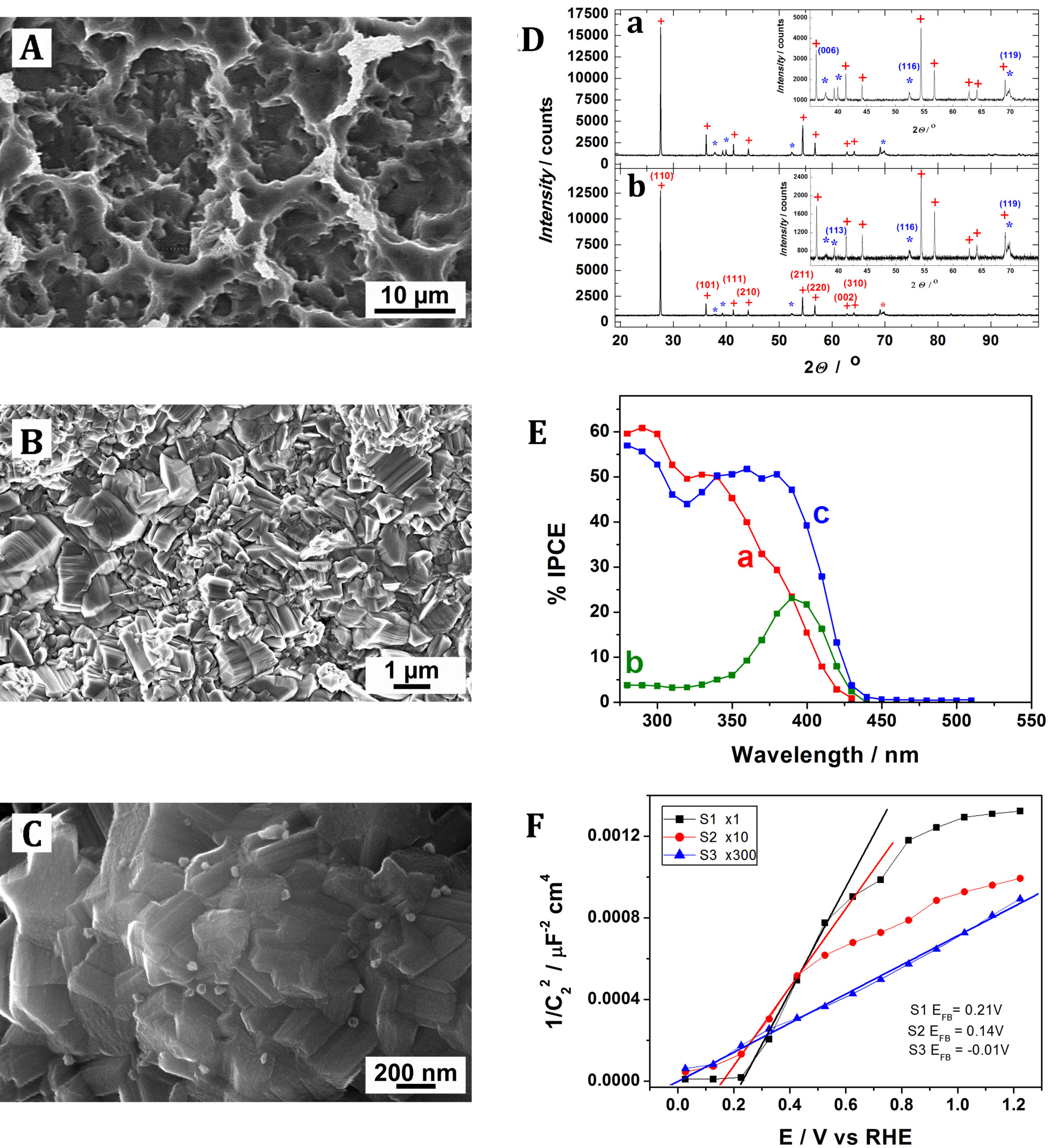
Figure 8. Electron scanning micrographs of TiO2 samples prepared by titanium oxidation in oxygen flow in the oven A) at 600°C, B) at 850°C and C) at 850°C – subsequently etched for 45 min in boiling KHC2O4. D) X-ray diffraction patterns of TiO2 samples prepared by Ti oxidation for 10 min at 850°C in oxygen flow after (a) and before etching (b) for 45 min in boiling KHC2O4. Addition signs denote reflections corresponding to rutile phase while asterisks denote those corresponding to the Ti3O phase. Inset shows magnified region between 35 and 70°. E) Spectral photoresponses of electrodes prepared by Ti oxidation in oxygen flow at 600°C (a), 850°C (b) and 850°C and subsequently etched for 45 min in boiling KHC2O4 (c). Curves recorded in 1M HClO4. F) Mott-Schottky plots for the rutile electrodes prepared by Ti oxidation at 600°C (S1), 850°C (S2) and at 850°C and subsequently etched in KHC2O4 (S3). Over the final period of the project we came back to, the earlier briefly investigated in our laboratory, idea of the seawater photo-electrolysis. Those investigations demonstrated remarkable stability of WO3 photo-anodes in the presence of chlorine being evolved from acidic chloride electrolytes. In a recent work, we designed relatively thick (up to 3-μm-thick) WO3 films, exhibiting large porosity across the whole film, that reconcile optimum light absorption with large (internal)photo-active surface area accessible to seawater electrolyte. This approach enabled reaching very high photo-currents, above 4.5 mA cm-2 , under simulated 1 sun AM 1.5G irradiation and at anodic potentials 0.4 V lower than thermodynamic Cl2 evolution potential. The chlorine was, in fact, the dominant product formed at the photo-anode. The operation of the seawater splitting cell was greatly simplified by the use of a palladium cathode that stored formed hydrogen as a hydride. The charged (Pd or mixed metal alloy) hydride electrode, removed from the PEC cell, might be potentially used as an anode in a hydride-oxygen battery (Adv. Energy Mater. 2019, 10, 1903213).
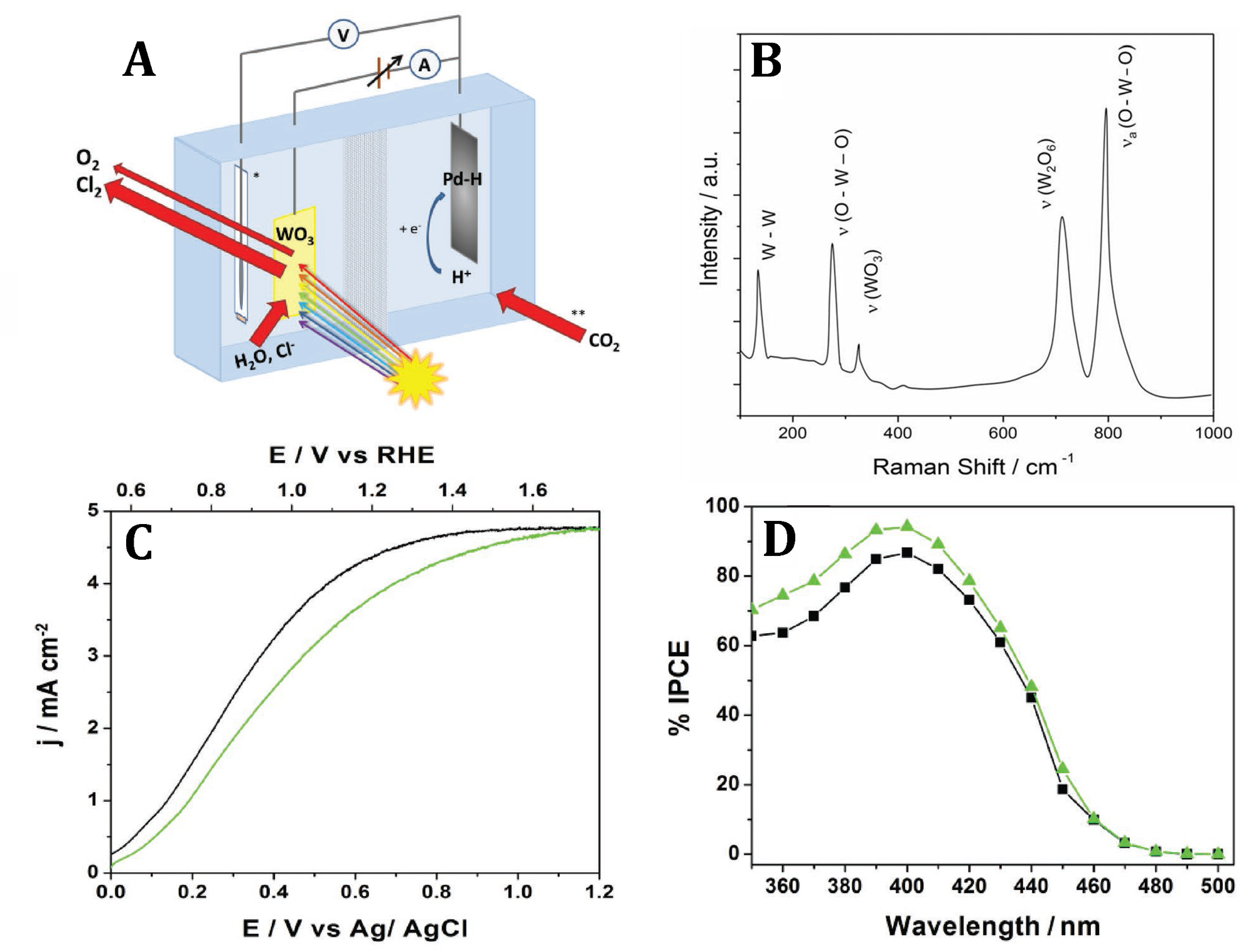
Figure 9. A) Schematic diagram of the seawater splitting PEC cell. The current of CO2 gas passing through the seawater represents a possible option for maintaining slightly acidic pH in the cathodic cell compartment. B) Raman spectrum of the WO3 film annealed at 550°C. C) Photo-anodic current vs potential (j-E) plots for a ~3 µm-thick WO3 electrode recorded in synthetic seawater under AM 1.5G (100 mW cm-2) illumination from the electrolyte side (black curve) and from the back side – through the FTO substrate (green trace). We note that the latter results have not been corrected for the light losses within the FTO substrate. D) Corresponding IPCE spectra; the IPCE values recorded under back side illumination (green plot) were corrected for the light absorption/reflection by the FTO layer on glass. List of publications:
|
|
|
Laboratory for Photoelectrochemistry and Solar Energy Conversion |
|